Chapter 15: Molecular Dynamics with GROMACS
This chapter provides a brief overview of using the molecular dynamics simulations with GROMACS, starting from PDB file manipulation. For detailed instructions and the latest features, please consult the official : GROMACS USER MANUAL .
15.1 Introduction
Molecular Dynamics (MD) simulations act as a computational microscope, allowing researchers to observe the atomic motions and thermodynamic behavior of biomolecules over time. By numerically solving Newton’s equations of motion, MD simulations provide insight into the structure, dynamics, and interactions of biological macromolecules under realistic physical conditions.
In this chapter, we present a step-by-step guide for performing MD simulations using GROMACS. The workflow begins with a small protein solvated in water and introduces the essential stages of a typical MD simulation pipeline. The simulation process includes several key steps:
- System Prepration: Generate molecular topologies and define the simulation box.
- Solvation and Ionization: Add water molecules and ions to mimic physiological conditions and neutralize the system.
- Equilibration: Stabilize the system through: i. Energy minimization to remove steric clashes. ii. NVT ensemble (constant number of particles, volume, and temperature) to stabilize temperature. iii. NPT ensemble (constant number of particles, pressure, and temperature) to stabilize pressure and system density.
- Production MD: Run the full molecular dynamics simulation to generate trajectories that capture the natural motion of the system.
Once the basic workflow is established, the same methodology can be extended to more complex biological systems, including:
- Protein–Ligand Complexes – to study molecular binding interactions relevant to drug discovery.
- Membrane Proteins – to simulate proteins embedded in lipid bilayers and investigate processes such as transport and signaling.
The value of an MD simulation lies in the analysis of the generated trajectories. This chapter introduces several core analysis tools available in GROMACS, including:
- Root Mean Square Deviation (RMSD) to evaluate structural stability.
- Radius of gyration (\(R_g\)) to measure protein compactness.
- Hydrogen bond analysis to examine intermolecular interactions.
These analyses help interpret simulation results and assess the structural stability, conformational changes, and interaction behavior of biomolecular systems.
15.2 Molecular Dynamics of Protein
In this section, we will perform a molecular dynamics simulation of hen egg white lysozyme (PDB ID: 1AKI) in an aqueous environment.
- Download the Structure: Download the protein structure from the RCSB Protein Data Bank.
- Fixing the Structure: Before starting the simulation, the protein structure should be checked and corrected for potential issues such as missing atoms, incomplete residues, or broken bonds. These problems are common in experimentally determined structures and may cause errors during topology generation.
For this step, we will use the online platform: BioExcel Buidling Blocks Workflows (BioBB-WFS)
This tool allows users to inspect and repair structural problems in PDB files before running molecular dynamics simulations.
After fixing the structure, the save the corrected PDB file as 1AKI_clean.pdb.
Why Structure Fixing is Important:
Missing or incorrect information in a PDB file can cause failures during topology generation using the GROMACS commandpdb2gmx. Common issues that may causepdb2gmxto fail include:
- Missing atoms: Residues with missing backbone or side-chain atoms can lead to topology generation errors.
- Missing residues: Some regions of a protein may be unresolved in experimental structures. These are often indicated by “MISSING” remarks in the PDB file.
- Incorrect chain termination: Each protein chain should end with a
TERrecord to correctly define chain boundaries.
- Generate Molecular Topology Using pdb2gmx: The topology file serves as a blueprint for the simulation, defining atomic interactions, bonding parameters, and force field information.
- Limitation of
pdb2gmx:
- Not fully automated: It cannot generate topologies for arbitrary molecules (e.g., ligands or complex cofactors).
- Restricted residue support: Works primarily with standard biomolecules (proteins, nucleic acids, some cofactors like NAD(H), ATP).
- Missing atoms/residues must be fixed before running pdb2gmx.
After removing water, verify that all necessary atoms are present in the PDB file before proceeding. Next, generate the molecular topology and structure file using the GROMACS pdb2gmx module:
Upon execution of the following commands, GROMACS will prompt you to choose a force field. Select: 15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
gmx pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water spce- After that will remove the crystal water molecules (HOH) using the command-line tools, text editors, or software like chimeraX. Avoid using Microsoft word processing software, as it may alter the file format.
grep -v HOH 1aki.pdb > 1AKI_clean.pdb- To remove water molecules using the command line.
Or we can specific the output of topol and posre file name with following commands.
gmx pdb2gmx -f 1AKI_clean.pdb -p topol.top -i posre.itp -o 1AKI_processed.gro -water spce .
Running
pdb2gmxcreates three key file:
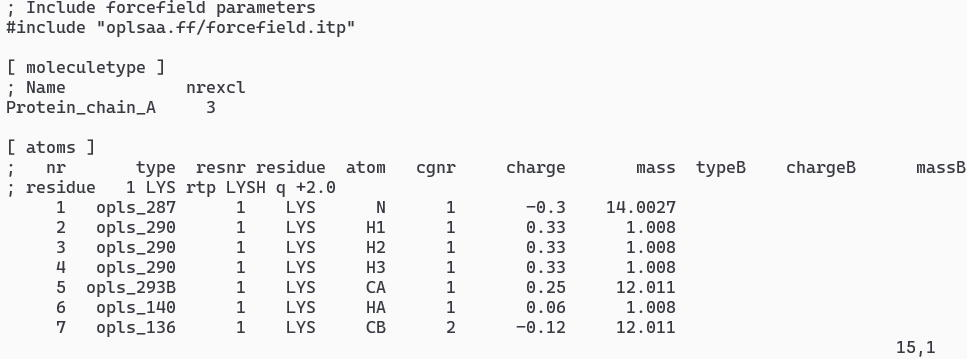
- Topology file (
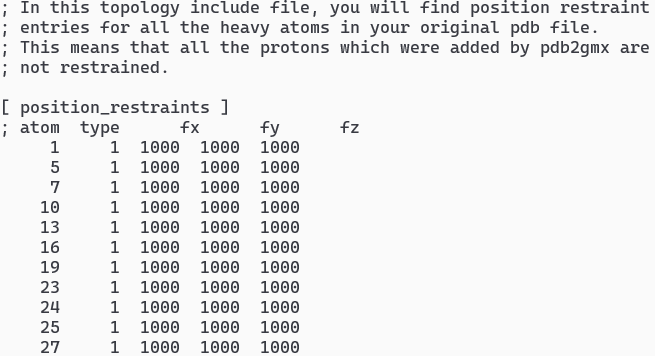
.top): Defines molecules, atoms , force field parameters. It contains nonbonded (atom types, charges) and bonded parameters (bonds, angles, dihedrals). Defines molecular interactions according to the chosen force field.- Position restraint file (
posre.itp): Used for restraining atoms (typically heavy atoms) during equilibration to prevent large movements.- Processed structure file (
.gro): (A GROMACS) structure file with atomic positions and velocities. The.grofiles is not mandatory, GROMACS supports multiple formats, including PDB (.pdb). If you prefer using the PDB format, simply specify the output filename with apdbextension.
Information about the water model can be found at Water Sctructure and Science .
Examine the Topology and restraints file using text file. You can find various comments and sections [atoms], [bonds], [pairs], [angles], and [dihedrals].
vi totp.top- Typical output of topology is shown in Figure 1vi posre.itp- Typical output of restrain is shown in Figure 1
1.3.2 Common Water Models in GROMACS
Depending on your installed force fields, the following water models are typically available in GROMACS:
ls /usr/local/gromacs/share/gromacs/top/- list available water models in the force field directory. Inside this directory, the.itpfiles (e.g.,spce.itp,tip3p.itp) correspond to available water models.
topology file must match molecular order with
.gro. The names list must match the[ moleculetype ]name for each species, not residue names or anything else.
- Create Box and Solvate: We will follow two steps There are two steps: i. defining the box dimensions using
gmx editconfmodule: Highly recommend the rhombic dodecahedron, as its volume ~71% of cubic box of same periodic distance, thus saving on number of water molecules that need to be added to solvate the protein.
gmx editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.0 -bt cubic
filling the box using solvate module (formerly called genbox): -c: center molecule in box, and -d 1.0: distance between solute and box (at least 1.0 nm from box), -bt cubic: cubix box type. We are using the periodic boundary conditions and protein should never see its periodic image.
ii. We will fill the box with solvent (water) using gmx solvate
gmx solvate -cp 1AKI_newbox.gro -cs spc216.gro -o 1AKI_solv.gro -p topol.top
-cp: configuration of protein. Filling box with solvent (water). The default solvent is Simple Point Charge water (SPC), with coordinates from spc216.gro, a generic equilibrated 3-point solvent model. We can also use spc216.gro as solvent configuration for SPC, SPC/E, or TIP3P, all are 3-point water models. Keep track of how many water molecules it has added. Note the changes to the [ molecules] directive of topol.top at the end of the file:
[ molecules ] ; Compound #mols Protein_A 1 SOL 10832
Note that if you use any other (non-water) solvent, solvate will not make these changes to your topology! Its compatibility with updating water molecules is hard-coded. For more information, gmx solvate -h in the command line.
- Energy minimization
- Need visualization xmgrace which can be install by:
conda install -c conda-forge grace- Using conda-forge (recommended)sudo apt-get install grace- Istalling via your system package managerxmgrace- to launch emgrace
- Equilibration in GROMACS Both NVT and NPT equilibration steps are essential in GROMACS (and molecular dynamics in general) to prepare your system for a stable production MD run.
- Step 1: NVT (Constant Number of particles, Volume, and Temperature). The goal is to stabilize the temperature of the system. Here letting the water/ions settle around your molecule while gently heating everything up. The procedue is:
- Keeps volume fixed.
- Gradually brings the system to the desired temperature (e.g., 300 K).
- Removes artifacts from energy minimization (e.g., unrealistic velocities).
- Applies position restraints to heavy atoms (usually protein) so the solvent can adapt around it. We let the protein, don’t wiggle too much while the solvent finds its groove.
The main reasons restrain Heavy Atoms (= all non-hydrogen atoms), Hydrogen atoms are light and fast-moving anyway, so they’re usually not restrained:
- Prevent large/unrealistic movements of solute atoms during the early stages of equilibration.
- Let the solvent (e.g., water, ions) adjust and relax around a stable solute.
- Avoid artificial distortion or unfolding of biomolecules while temperature or pressure is ramping up.
Especially right after energy minimization, some water or ions may be poorly placed. If the protein moves too freely, things can go wild — leading to simulation crashes or bad dynamics.
During production MD, we usually remove restraints, so the system evolves naturally. Restraints are only meant for the equilibration phase. In
.mdpfile, we includedefine = -DPOSRESwhich tells GROMACS to include the posre.itp file for atoms that are marked as restrained in your topology.
- Step 2: NPT (Constant Number of particles, Pressure, and Temperature). Goal: Stabilize the pressure and let the box size/volume adjust naturally. Here, we are letting the box “breathe” and find its natural size under realistic pressure. The procedue is:
- Keeps temperature constant (as already stabilized by NVT).
- Allows box volume to fluctuate so pressure can equilibrate (target: 1 bar).
- Ensures density and pressure of your system match experimental or physiological conditions.
- Analysis Step 1 Using
gmx trjconv(a pos-processing tool) to remove periodic boundary conditions (PBC) and center your molecule in the simulation box — a common step before visualization or analysis. Let’s break this command down and make sure you run it correctly.gmx trjconv -s md_0_1.tpr -f md_0_1.xtc -o md_0_1_noPBC.xtc -pbc mol -center
- Flags:
s md_0_1.tpr: Structure/topology file (needed for box info and atom groups).f md_0_1.xtc: Original trajectory file (with PBC effects).o md_0_1_noPBC.xtc: Output trajectory (with fixed PBC and centered).pbc mol: Treats molecules as whole units when unwrapping across PBC.center: Centers selected group in the box.
When we run this, GROMACS will ask twice:
- “Select group for centering:” Choose the group you’d like to center, usually something like: Protein, Backbone, Protein-H, or DNA (if applicable).
- “Select output group:” Choose the entire system or what you want to output: System, Protein (if you want only the protein), or Protein + solvent, etc.
gmx rms -s em.tpr -f md_0_1_noPBC.xtc -o rmsd_xtal.xvg -tu ns- calculating RMSD relative to the minimized structure (em.tpr), likely to compare how much your system deviates from the initial crystal or model structure. Solid move, especially if you’re interested in structural stability over time.s em.tpr: Reference structure — your energy-minimized (often crystallographic) structure.f md_0_1_noPBC.xtc: Trajectory with PBC artifacts removed and system centered.o rmsd_xtal.xvg: Output RMSD values over time.tu ns: Report time in nanoseconds (default is picoseconds).
You’ll be prompted twice:
Group for least-squares fit (alignment)
- e.g., Protein Group for RMSD calculation
- e.g., Protein